
USC Today Home Page
Top Stories from USC News

Honorees join more than 40 USC faculty as fellows of the American Association for the Advancement of Science.

USC scientists find that adolescents who grapple with the bigger meaning of social situations experience greater brain growth, which predicts stronger identity development and life satisfaction years later.

During Second Chance Month, USC Rossier professors are making meaningful contributions to criminal justice reform.

Wallis Annenberg Hall and USC Stage to feature faculty and students during two-day event on University Park Campus.

Assignment: Earth begins with us
Assignment: Earth is USC’s comprehensive sustainability framework, which guides the university in its mission to foster a healthy, just, and thriving campus and global community. It encompasses various initiatives and strategies to promote environmental stewardship, social equity and economic vitality within the university and beyond.
Celebrating Earth Month at USC
Celebrating Earth Month at USC includes events, initiatives and activities dedicated to raising awareness about environmental issues, promoting sustainability practices and inspiring action among the university community. Here are our latest stories.

Eco Film & Media Arts Festival, Urban Trees Initiative receive national sustainability accolades
USC programs are recognized for student engagement and racial equity from the Association for the Advancement of Sustainability in Higher Education.

Three USC schools collaborate to reimagine the L.A. River
Researchers in landscape architecture, fluid mechanics and augmented reality have created an interactive hydraulic model alongside the river.

USC Sea Grant research, community outreach show approval of aquaculture can be earned
People across all demographics could change their minds when they learned about the environmental benefits of seafood farming, USC research suggests.
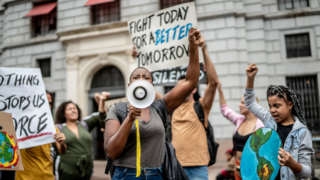
From throwing soup to suing governments, there’s strategy to climate activism’s seeming chaos
USC’s Shannon Gibson says that with international climate talks failing to make progress fast enough, activists are radically rethinking how to be most effective in the streets, political arenas and courtrooms.

Surprising discovery about coral’s resilience could help reefs survive climate change
USC Dornsife researchers studying a common Caribbean coral’s ability to adapt to rising temperatures turn up an unexpected result.

Looking to the Sky
Trojans gathered in Hahn Plaza on the University Park Campus on Monday for a lively eclipse viewing party. Learn from USC experts about the symbolism of the celestial rarity and more.
Stories from USC Trojan Family Magazine


Music and culture at the Los Angeles Memorial Coliseum

Learning through food at USC

Experts on trending topics



